Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome, often with a life-threatening course of disease. While in children with HLH in most cases an underlying mutation in immunoregulatory genes is detectable acquired HLH in adults is very heterogeneous with a variety of triggers and predispositions. For treating physicians it is often difficult to distinguish HLH from sepsis, SIRS or a severe infection. One reason for the high mortality in adult HLH is the delay of diagnosing the syndrome.
Pathophysiological HLH is based on an ineffective immune response with impaired pathogen elimination, which is caused by a mutation in genetic HLH resulting in dysfunction of cytotoxic T cells. The term hemophagocytic lymphohistiocytosis developed because of the increased hemophagocytic activity of macrophages and the cytokine-induced massive lymphoproliferation. Clinical signs of patients may be hepatosplenomegaly, lymphadenopathy, jaundice, purpura and an increasing cytopenia (anemia and extreme leukopenia). Biopsy or autopsy shows phagocytic macrophages that can be located generalized in the lymphatic tissue (Hemophagocytosis; in bone marrow, spleen or lymph nodes). Moreover, an involvement of the skin, kidneys, gastrointestinal tract or central nervous system is possible.
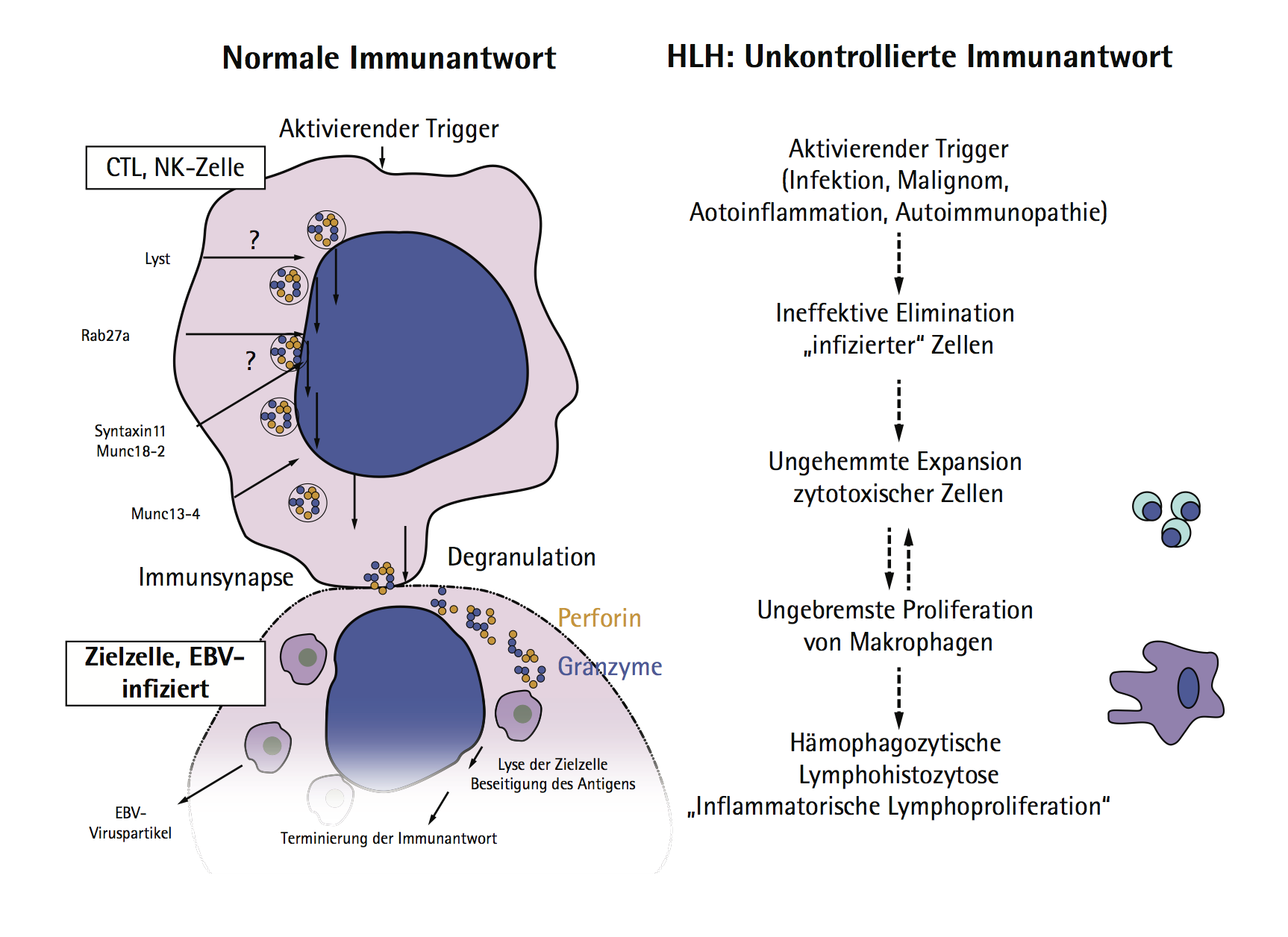
Pathophysiology of HLH.
A variety of synonyms for the clinical syndrome were used historically in the literature (see Table 1). The term used today was first introduced by Risdall et al. (virus-associated Hemophagocytic syndrome). HLH in the context of autoimmune diseases (e.g. M. Still) is also known as macrophage activation syndrome.
| author | team |
| Bykowa et al 1928 | Systemretikulose (German publication) |
| Scott and Robb-Smith et al 1939 | Histiocytic Medullary Reticulosis |
| Farquahr et al 1952 | Familial Hemophagocytic Reticulosis |
| Risdall et al 1979 | Hemophagocytic Lymphohistiocytosis (HLH) |
Table 1: Historical used HLH terms
Due to the HLH-mutations that have been described in pediatric patients in the last 20 years, HLH was historically classified into a primary (genetic) form in children and a secondary form in adults, which is caused by infectious, malignant or autoimmune trigger. The strict division in primary and secondary HLH seems to be obsolete because in the literature many case reports of adults with HLH and an underlying genetic defect appear (in some cases up to the 6th decade of life). On the other hand our increased understanding of the pathophysiology shows that even in genetic HLH in childhood an infectious trigger mechanism is needed to trigger the clinical picture. The presently preferred nomenclature is a classification into hereditary and acquired HLH..