Die Diagnose einer HLH kann aufgrund der Überlappung mit anderen Hyperinflammationssyndromen (SIRS , Sepsis) schwierig sein. Die Diagnosekriterien der HLH wurden bislang nur für pädiatrische Patienten etabliert. Daher ist für die Diagnosestellung immer auch die Dynamik der einzelnen Parameter und die Erfahrung des behandelnden Arztes entscheidend. Der Nachweis von Hämophagozytose beweist nicht das Vorliegen einer HLH und ist für die Diagnosestellung auch nicht zwingend notwendig. Es sollte bei kritisch kranken Patienten und unklaren Fällen unbedingt eines der HLH-Zentren in Jena oder Hamburg kontaktiert werden.
In Tabelle 2 sind die derzeit verwendeten Diagnosekriterien (HLH Study Group der Histiocyte Society, Henter et al 2007) aufgeführt. Einen Diagnosepfad, welcher der aktuellen Onkopedia-Leitlinie entnommen ist, zeigt Abbildung 2.
- Familiäre Erkrankung/bekannter genetischer Defekt
oder
- 5 von 8 der typischen Kriterien erfüllt:
| Fieber |
| Splenomegalie |
| Mindestens Bi-Zytopenie (Hb <9 g/dl; Thrombozyten <100/nl; Neutrophile <1/nl) |
| Hypertriglyceridämie (≥3 mmol/l) und/oder Hypofibrinogenämie (<1.5g/l) |
| Ferritin ≥500 μg/l |
| sCD25 ≥2,400 U/ml (sCD25 = löslicher IL-2-Rezeptor) |
| Verminderte oder fehlende NK-Zell-Aktivität |
| Hämophagozytose (Knochenmark, Milz, Lymphknoten) |
- Unterstützende Kriterien: neurologische Symptome, Erhöhung von LDH, Transaminasen, Bilirubin (Verschlussikterus)
Tabelle 2: HLH-Diagnosekriterien
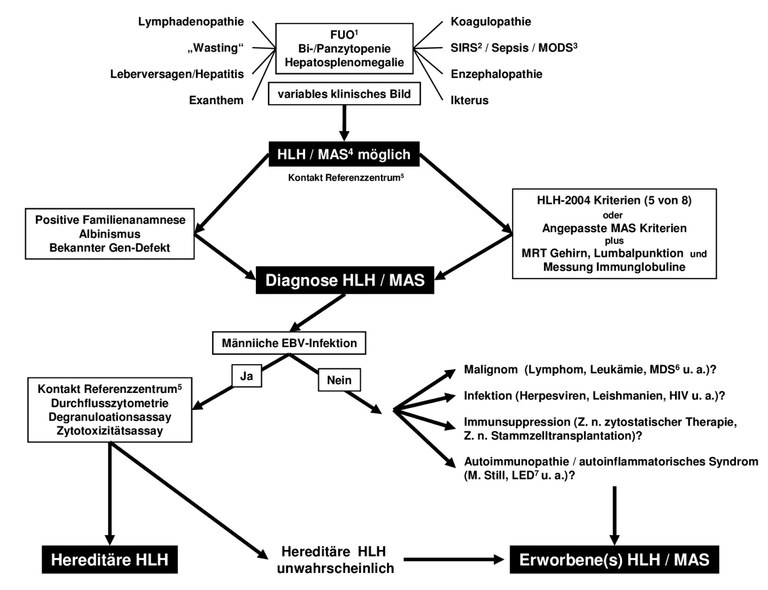
Abbildung 2: Diagnosepfad bei Verdacht auf HLH (aus: Onkopedia Leitlinie zur HLH, Onkopedia)
Legende: 1FUO – Fieber unklarer Genese (Fever of Unknown Origin); 2SIRS – Systemisches Inflammatorisches Response-Syndrom; 3MODS – multiples Organversagen (Multiple Organ Dysfunction Syndrome); 4MAS – Macrophase Activation Syndrome; 5Referenzzentrum Pädiatrie: http://www.uke.de/kliniken/haematologie/index_39115.php; Referenzzentrum Erwachsene Patienten: http://www.hlh-registry.org; 6MDS – Myelodysplastisches Syndrom; 7LED – Lupus Erythematodes Disseminatus