Die Hämophagozytische Lymphohistiozytose (HLH) ist ein hyperinflammatorisches Syndrom mit einem häufig lebensbedrohlichen Verlauf. Während bei Kindern mit HLH in den meisten Fällen eine Mutation in immunregulierenden Genen zugrundeliegt, zeigt sich die erworbene HLH bei Erwachsenen sehr heterogen mit einer Vielzahl unterschiedlicher Auslöser und Prädispositionen. Differenzialdiagnostisch ist es dann nur schwer von einer Sepsis, einem SIRS bzw. einer schweren Infektion zu unterscheiden. Nicht zuletzt aufgrund von Diagnoseverzögerungen ist die Mortalität bei adulter HLH sehr hoch. Pathophysiologisch liegt dem Syndrom eine ineffektive Immunantwort mit gestörter Pathogenelimination zugrunde, die bei genetischen Formen durch eine mutationsbedingte Dysfunktion der zytotoxischen T-Zellen bedingt ist. Die gesteigerte Hämophagozytose-Aktivität der Makrophagen sowie die durch massive unkontrollierte Zytokinausschüttung bedingte Lymphoproliferation waren namengebend für das Syndrom. Klinische Zeichen der Patienten können Hepatosplenomegalie, Lymphadenopathie, Ikterus, Purpura und eine zunehmende Zytopenie (Anämie und extreme Leukopenie) sein. Bioptisch oder autoptisch können sich generalisiert im lymphatischen Gewebe phagozytierende Makrophagen befinden (Hämophagozytose; in Knochenmark, Milz oder Lymphknoten). Auch eine Beteiligung von Haut, Nieren, Gastrointestinaltrakt oder ZNS ist möglich.
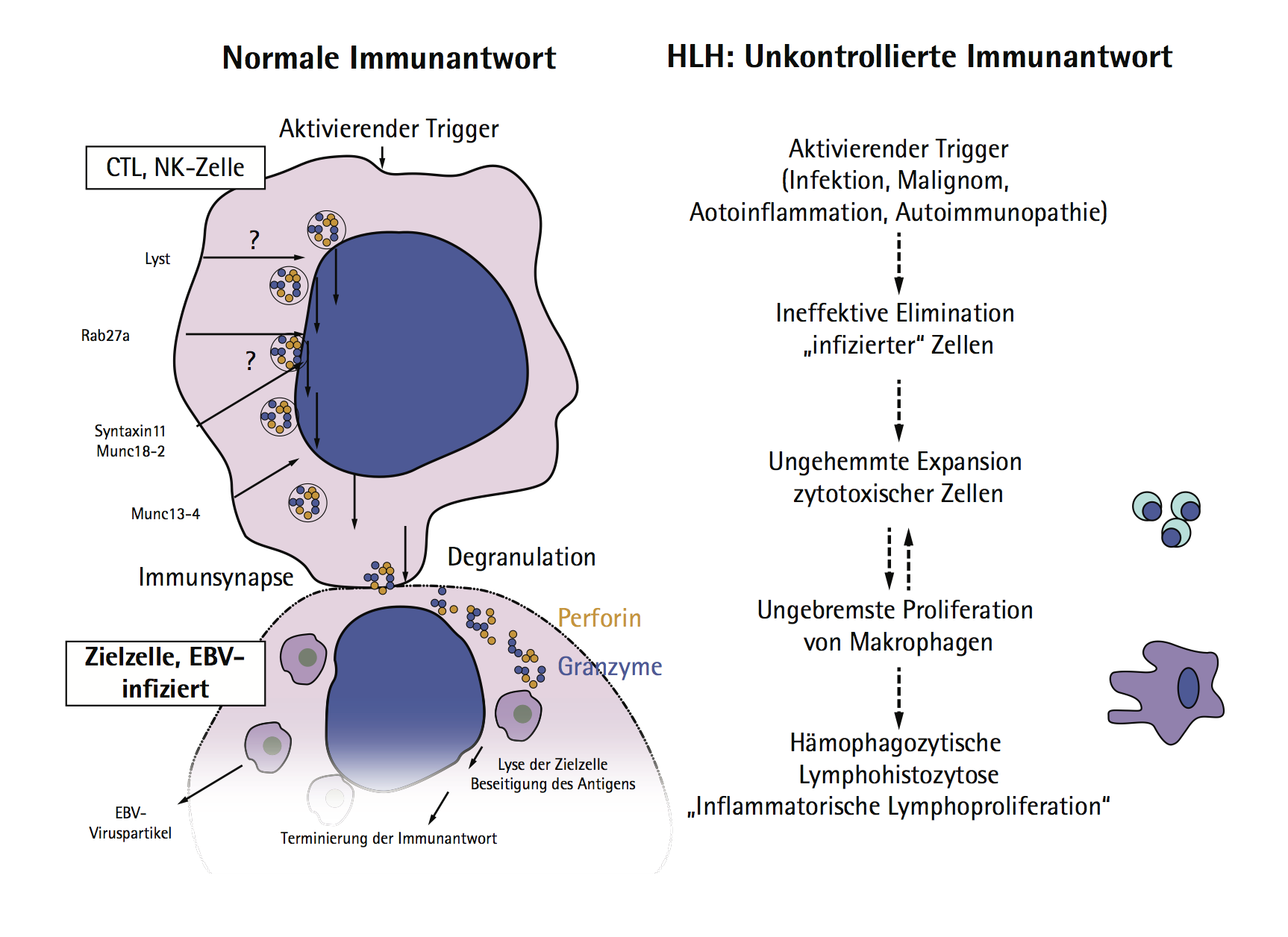
Abbildung: Pathophysiologie der HLH.
Historisch bedingt werden in der Fachliteratur eine Vielzahl von Synonymen für das Krankheitssyndrom verwendet, was zur Begriffsverwirrung beiträgt (siehe Tabelle 1). Der bis heute verwendete Begriff der Hämophagozytischen Lymphohistiozytose (HLH) wurde erstmals von Risdall et al als virusassoziiertes hämophagozytisches Syndrom eingeführt. Eine im Kontext von Autoimmunerkrankungen aufgetretene HLH (z.B. bei M. Still) wird auch als Makrophagenaktivierungs-Syndrom bezeichnet.
| Autor | Begriff |
| Bykowa et al 1928 | Systemretikulose |
| Scott und Robb-Smith et al 1939 | Histiozytische Medulläre Retikulose |
| Farquahr et al 1952 | Familiäre Hämophagozytische Retikulose |
| Risdall et al 1979 | Hämophagozytische Lymphohistiozytose (HLH) |
Tabelle 1: Historisch verwendete HLH-Begriffe
Aufgrund der in den letzten 20 Jahren beschriebenen HLH-Mutationen bei pädiatrischen Patienten erfolgte historisch die Einteilung in eine primäre, genetische Form bei Kindern und die sekundäre HLH bei Erwachsenen, die durch infektiöse, maligne oder autoimmune Trigger hervorgerufen wird. Die strikte Trennung in primäre und sekundäre HLH scheint überholt zu sein, da zunehmend Fallberichte von Erwachsenen mit HLH und zugrundeliegendem genetischen Defekt (teilweise bis in die 6. Lebensdekade) beschrieben werden. Andererseits zeigt unser zunehmendes Verständnis der Pathophysiologie, dass auch bei der genetischen HLH im Kindesalter meist ein infektiöser Triggermechanismus notwendig zur Auslösung des klinischen Bildes ist. Die derzeit bevorzugte Nomenklatur ist eine Einteilung in hereditäre und erworbene HLH.